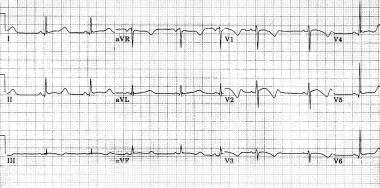
Síndrome de Jervell y Lange-Nielsen
El síndrome de QT largo es una enfermedad hereditaria arritmogénica. Existen dos formas: el síndrome autosómico dominante Romano-Ward y la forma autosómica recesiva Jervell Lange-Nielsen, síndrome asociado con sordera neuro-sensorial.
El primer caso descrito por Anton Jervell y Fred Lange Nielsen en 1957
En el síndrome de Jervell la incidencia estimada es de 1,6-6 casos por millón. La prolongación del intervalo QT se asocia con taquiarritmias torsade de pointes, incluyendo taquicardia ventricular y fibrilación ventricular, que puede terminar con síncope o muerte súbita. La anemia y niveles elevados de gastrina también son características frecuentes de dicho síndrome.
La presentación clásica es un niño sordo que experimenta episodios sincópales recurrentes durante períodos de estrés, el ejercicio o el miedo, convulsiones o muerte súbita. El cincuenta por ciento de las personas con con síndrome de Jervell han presentado eventos cardíacos antes de la edad de tres años.
El diagnóstico se establece en un niño con sordera congénita neurosensorial, intervalo QT largo, y la presencia de mutaciones heterocigotas u homocigotas en los genes KCNQ1 o KCNE1, genes que codifican para canales de sodio y potasio.